kraken源码分析
contact me

或者用邮件交流 jacky.wucheng@foxmail.com
前言
大家都知道Kraken是一个序列分类器软件,经常被用来对序列进行物种鉴定。我们公司叫做 量化健康,主要从事于人体肠道微生物的研究,我们用Kraken来进行微生物“种”水平的分类鉴定。
研究Kraken的起因是我想弄明白这几个问题
- 为什么Kraken的分析速度那么快?
- 为什么kraken的数据库有几百G那么大?
- 为什么Kraken建库的速度非常慢?
- 为什么Kraken数据库的载入速度非常慢?
- Kraken的数据库能否拆分使得其能够分布式运行?
分析
我们参照着 Kraken-Manual的顺序来分析。
Kraken目录结构很简单
.
├── CHANGELOG
├── LICENSE
├── README.md
├── docs
├── install_kraken.sh # 安装脚本
├── scripts # 一系列shell或者perl脚本用来做胶水,包含了所有非计算密集型代码
└── src # c++写成的核心功能,包含了所有计算密集型代码
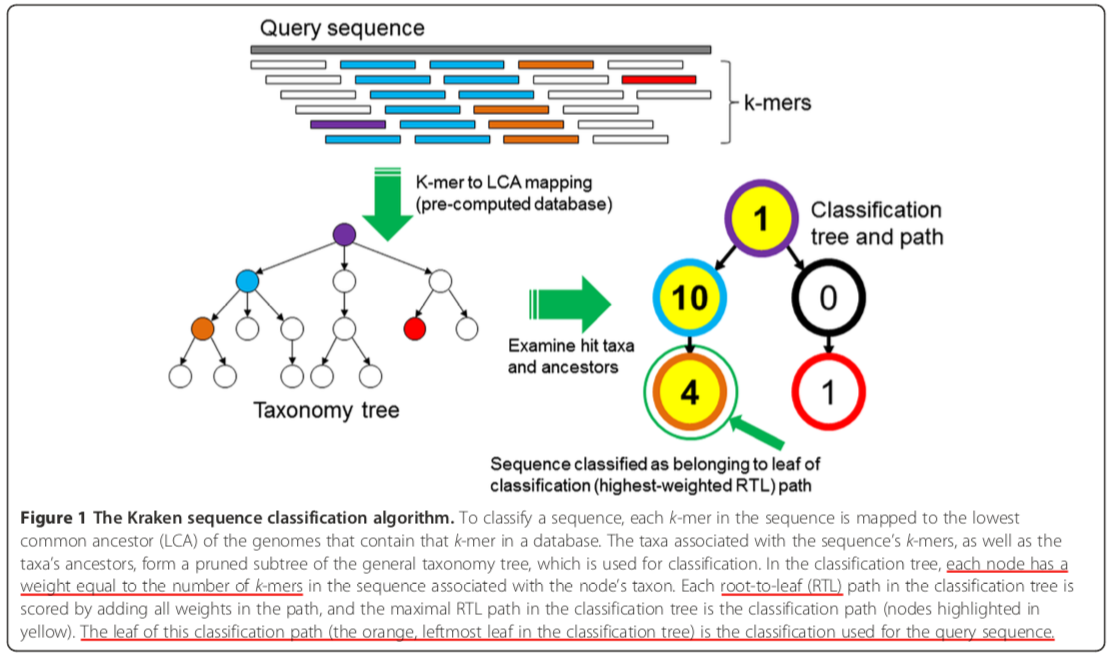
原理
Kraken的基本原理是:
- 从NCBI上下载所需物种的序列和分类数据,然后做成k-mer对应taxon的数据库。
- 将数据库和索引文件mmap到内存,将待鉴定序列切成k-mer,比对到数据库上获得其lca_taxon以及比对上的次数。
- 将上述的
taxon: count的数据构建成下述classification tree,然后计算每条root-to-leaf上的所有权重和,最大者即为该条序列的分类树。
软件安装
软件安装由install_kraken.sh脚本实现。核心就是接收一个参数:“安装路径”,然后进入src目录进行make,将二进制文件copy到安装路径中而已。
数据来源和数据格式
数据全部来源于NCBI。
taxonomy文件列表
wget $FTP_SERVER/pub/taxonomy/taxdump.tar.gz
wget $FTP_SERVER/pub/taxonomy/accession2taxid/nucl_est.accession2taxid.gz
wget $FTP_SERVER/pub/taxonomy/accession2taxid/nucl_gb.accession2taxid.gz
wget $FTP_SERVER/pub/taxonomy/accession2taxid/nucl_gss.accession2taxid.gz
wget $FTP_SERVER/pub/taxonomy/accession2taxid/nucl_wgs.accession2taxid.gz
taxonomy/nucl_gb.accession2taxid 文件内容格式
{
'accession': 'A00002',
'accession.version': 'A00002.1',
'gi': '2',
'taxid': '9913'
}
taxdump.tar.gz 压缩文件列表
-rw-r--r-- tm/tm 16865033 2018-06-06 07:20 citations.dmp
-rw-r--r-- tm/tm 3766694 2018-06-06 07:20 delnodes.dmp
-rw-r--r-- tm/tm 442 2018-06-06 07:20 division.dmp
-rw-r--r-- tm/tm 15188 2018-06-06 07:20 gc.prt
-rw-r--r-- tm/tm 4575 2018-06-06 07:20 gencode.dmp
-rw-r--r-- tm/tm 923141 2018-06-06 07:20 merged.dmp
-rw-r--r-- tm/tm 155543951 2018-06-06 07:20 names.dmp
-rw-r--r-- tm/tm 120511689 2018-06-06 07:20 nodes.dmp
-rw-rw---- domrach/tm 2652 2006-06-13 19:04 readme.txt
nodes.dmp文件内容格式
{'GenBank_hidden_flag': '0',
'comments': '',
'division_id': '8',
'embl_code': '',
'genetic_code_id': '1',
'hidden_subtree_root_flag': '0',
'inherited_GC_flag': '0',
'inherited_MGC_flag': '0',
'inherited_div_flag': '0',
'mitochondrial_genetic_code_id': '0',
'parent_tax_id': '1',
'rank': 'no rank',
'tax_id': '1'}
names.dmp文件内容格式
{'name class': 'synonym',
'name_txt': 'all',
'tax_id': '1',
'unique name': ''}
{'name class': 'scientific name',
'name_txt': 'root',
'tax_id': '1',
'unique name': ''}
library文件列表
kraken-build --db $KRAKEN_DB_NAME --download-library archaea
kraken-build --db $KRAKEN_DB_NAME --download-library bacteria
kraken-build --db $KRAKEN_DB_NAME --download-library viral
bacteria/assembly_summary.txt文件内容格式
{' assembly_accession': 'GCF_000010525.1',
'asm_name': 'ASM1052v1',
'assembly_level': 'Complete Genome',
'bioproject': 'PRJNA224116',
'biosample': 'SAMD00060925',
'excluded_from_refseq': '',
'ftp_path': 'ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/010/525/GCF_000010525.1_ASM1052v1',
'gbrs_paired_asm': 'GCA_000010525.1',
'genome_rep': 'Full',
'infraspecific_name': 'strain=ORS 571',
'isolate': '',
'organism_name': 'Azorhizobium caulinodans ORS 571',
'paired_asm_comp': 'identical',
'refseq_category': 'representative genome',
'relation_to_type_material': 'assembly from type material',
'release_type': 'Major',
'seq_rel_date': '2007/10/16',
'species_taxid': '7',
'submitter': 'University of Tokyo',
'taxid': '438753',
'version_status': 'latest',
'wgs_master': ''}
建库
Kraken的数据库至少包含了如下4个文件
database.kdb: Contains the k-mer to taxon mappingsdatabase.idx: Contains minimizer offset locations in database.kdbtaxonomy/nodes.dmp: Taxonomy tree structure + rankstaxonomy/names.dmp: Taxonomy names
建库的入口在scripts/kraken-build中。这是个邪恶的perl脚本。他根据输入参数的不同,调用不同的shell去完成功能。
如这条指令
kraken-build --standard --db $DBNAME
实际上是调用了standard_installation.sh这个shell,见
sub standard_installation {
exec "standard_installation.sh";
}
去NCBI_SERVER="ftp.ncbi.nlm.nih.gov"下载taxonomy和archaea,bacteria,viral数据。
分别查看download_taxonomy.sh和download_genomic_library.sh脚本了解下载逻辑。
check_for_jellyfish.sh
kraken-build --db $KRAKEN_DB_NAME --download-taxonomy
kraken-build --db $KRAKEN_DB_NAME --download-library archaea
kraken-build --db $KRAKEN_DB_NAME --download-library bacteria
kraken-build --db $KRAKEN_DB_NAME --download-library viral
kraken-build --db $KRAKEN_DB_NAME --build --threads $KRAKEN_THREAD_CT \
--jellyfish-hash-size "$KRAKEN_HASH_SIZE" \
--max-db-size "$KRAKEN_MAX_DB_SIZE" \
--minimizer-len $KRAKEN_MINIMIZER_LEN \
--kmer-len $KRAKEN_KMER_LEN \
$WOD_FLAG
下载完毕之后就利用scripts/build_kraken_db.sh该脚本进行建库。
这是kraken-build --standard --db $DBNAME命令运行第二遍输出的结果(第一遍是新建库,跟下载速度相关,可能要1,2天时间):
Found jellyfish v1.1.11
Skipping archaea, already completed library download
Skipping bacteria, already completed library download
Skipping viral, already completed library download
Kraken build set to minimize disk writes.
Skipping step 1, k-mer set already exists.
Skipping step 2, no database reduction requested.
Skipping step 3, k-mer set already sorted.
Skipping step 4, GI number to seqID map now obsolete.
Skipping step 5, seqID to taxID map already complete.
Skipping step 6, LCAs already set.
step-1
利用jellyfish软件切k-mer,jellyfish的使用方法可以参考Jellyfish详解。
jellyfish count -m $KRAKEN_KMER_LEN -s $KRAKEN_HASH_SIZE -C -t $KRAKEN_THREAD_CT \
-o database /dev/fd/0
而且无论jellyfish产生出了几个数据库文件,都会被merge成一个。
# Merge only if necessary
if [ -e "database_1" ]
then
jellyfish merge -o database.jdb.tmp database_*
else
mv database_0 database.jdb.tmp
fi
自此,就生成了database.jdb文件,文件内容是 “k-mer :count”。
step-2
用来缩减数据库大小的。这里不展开了。
step-3
用来对database.jdb进行排序,并且生成索引文件的。
db_sort -z $MEMFLAG -t $KRAKEN_THREAD_CT -n $KRAKEN_MINIMIZER_LEN \
-d database.jdb -o database.kdb.tmp \
-i database.idx
# Once here, DB is sorted, can put file in proper place.
mv database.kdb.tmp database.kdb
排序逻辑在src/db_sort.cpp文件中。核心逻辑在bin_and_sort_data函数中:
static void bin_and_sort_data(KrakenDB &kdb, char *data, KrakenDBIndex &idx) {
......省略代码
// Create a copy of the offsets array for use as insertion positions
vector<uint64_t> pos(offsets, offsets + entries);
for (uint64_t i = 0; i < kdb.get_key_ct(); i++) {
input_file.read(pair, pair_size);
uint64_t kmer = 0;
memcpy(&kmer, pair, key_len);
uint64_t b_key = kdb.bin_key(kmer, nt);
char *pair_pos = data + pair_size * pos[b_key]++;
// Copy pair into correct bin (but not final position)
memcpy(pair_pos, pair, pair_size);
if (Zero_vals)
memset(pair_pos + key_len, 0, val_len);
}
input_file.close();
// Sort all bins
#pragma omp parallel for schedule(dynamic)
for (uint64_t i = 0; i < entries; i++) {
qsort(data + offsets[i] * pair_size,
offsets[i+1] - offsets[i], pair_size,
pair_cmp);
}
}
我们看到这里要进行一个qsort快速排序,快排的平均复杂度是O(nlogn),最坏情况是O(n^2^),对于将近200G的数据在内存里进行快排,速度慢也是理所应当的,这步其实就是建库过程中除了下载数据库步骤外最耗时的地方。
关于索引, 参考Kraken-Paper
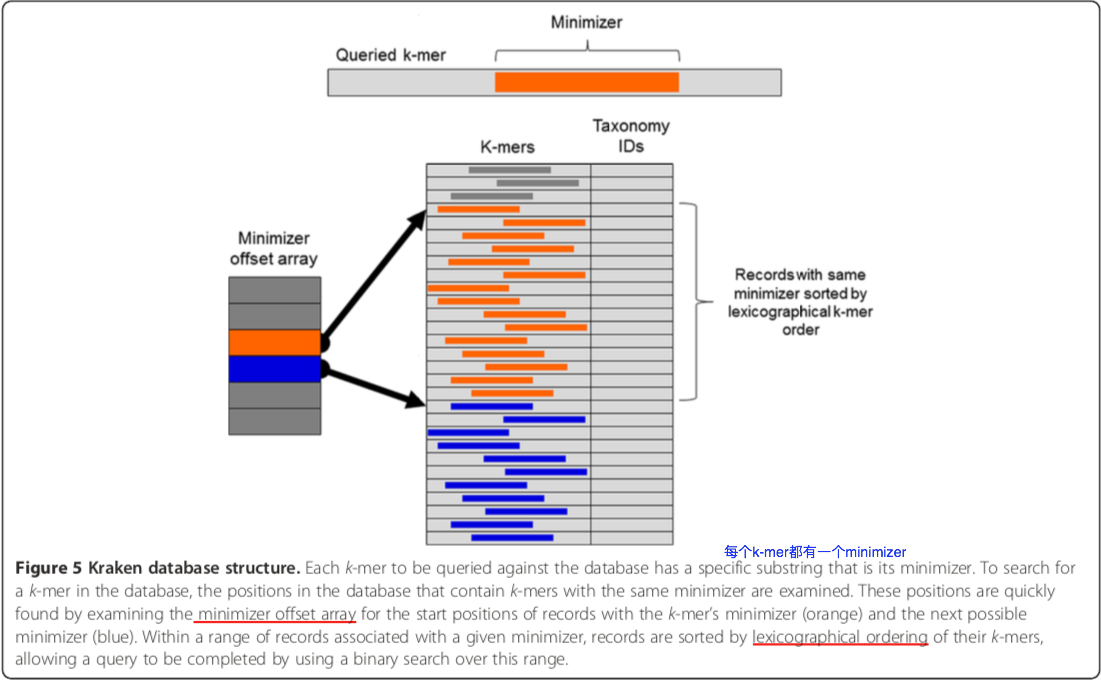
因为k-mer本身切割的性质使得相邻的k-mer都含有一个相同的子串,叫做minimizer,所以使用minimizer对排好序的k-mer进行分组做索引,见上图。这使得后续的查询操作效率很高。
另外,因为设置了Zero_vals标记,jellyfish产生的 k-mer:count的pair,其count部分已经全部被置为0,用来存储后续将会用到的LCA_taxon。
该步骤最终产生了database.kdb和database.idx这两个文件。
step-4
该步骤已经被废弃了,跳过。
step-5
将seqID跟taxID做映射。目的就是为了将待鉴定序列的k-mer比对到数据库里获取其taxonID。这由邪恶的perl+shell脚本来实现:
find library/ -maxdepth 2 -name prelim_map.txt | xargs cat > taxonomy/prelim_map.txt
if [ ! -s "taxonomy/prelim_map.txt" ]; then
echo "No preliminary seqid/taxid mapping files found, aborting."
exit 1
fi
grep "^TAXID" taxonomy/prelim_map.txt | cut -f 2- > $seqid2taxid_map_file.tmp || true
if grep "^ACCNUM" taxonomy/prelim_map.txt | cut -f 2- > accmap_file.tmp; then
if compgen -G "taxonomy/*.accession2taxid" > /dev/null; then
lookup_accession_numbers.pl accmap_file.tmp taxonomy/*.accession2taxid > seqid2taxid_acc.tmp
cat seqid2taxid_acc.tmp >> $seqid2taxid_map_file.tmp
rm seqid2taxid_acc.tmp
else
echo "Accession to taxid map files are required to build this DB."
echo "Run 'kraken-build --db $KRAKEN_DB_NAME --download-taxonomy' again?"
exit 1
fi
fi
mv $seqid2taxid_map_file.tmp $seqid2taxid_map_file
line_ct=$(wc -l $seqid2taxid_map_file | awk '{print $1}')
echo "$line_ct sequences mapped to taxa. [$(report_time_elapsed $start_time1)]"
fi
我们只要了解到其最终产生了seqid2taxid.map这个文件,内容为
kraken:taxid|1980463|NC_034515.1 1980463
kraken:taxid|1980463|NC_034525.1 1980463
kraken:taxid|1980463|NC_034528.1 1980463
后续的流程里会利用该文件在内存里构建出map<uint32_t, uint32_t> Parent_map;这样的一个数据结构,用map的方式行成了一棵“界门纲目科属种”的分类树。细节在后文中讲。
step-6
对database.kdb文件进行set LCA操作。
核心逻辑在set_lcas.cpp文件中
void process_single_file() {
......省略代码
while (reader.is_valid()) {
dna = reader.next_sequence();
if (! reader.is_valid())
break;
uint32_t taxid = ID_to_taxon_map[dna.id];
if (taxid) {
#pragma omp parallel for schedule(dynamic)
for (size_t i = 0; i < dna.seq.size(); i += SKIP_LEN)
set_lcas(taxid, dna.seq, i, i + SKIP_LEN + Database.get_k() - 1);
}
if (isatty(fileno(stderr)))
cerr << "\rProcessed " << ++seqs_processed << " sequences";
else if (++seqs_processed % 500 == 0)
cerr << "Processed " << seqs_processed << " sequences.\n";
}
......省略代码
}
void set_lcas(uint32_t taxid, string &seq, size_t start, size_t finish) {
KmerScanner scanner(seq, start, finish);
uint64_t *kmer_ptr;
uint32_t *val_ptr;
while ((kmer_ptr = scanner.next_kmer()) != NULL) {
if (scanner.ambig_kmer())
continue;
val_ptr = Database.kmer_query(
Database.canonical_representation(*kmer_ptr)
);
if (val_ptr == NULL) {
if (! Allow_extra_kmers)
errx(EX_DATAERR, "kmer found in sequence that is not in database");
else
continue;
}
*val_ptr = lca(Parent_map, taxid, *val_ptr);
}
}
通过上面两个函数我们可以知道database.kdb最终被处理为k-mer: lca_taxon_id的数据结构。
最终database.kdb和database.idx两个文件被处理完毕。这一步其实也是非常耗时的。
鉴定
鉴定的命令为kraken --db $DBNAME seqs.fa。并且为了加速鉴定过程,可以使用preload指令将数据库和索引预先加载到内存里去:kraken --preload --db $DBNAME seqs.fa。
鉴定的命令最终使用到的逻辑在classify.cpp文件中,其中使用到了Populate_memory这个变量来标记要不要对数据库和索引进行preload。并且database.kdb和database.idx都是通过QuickFile这个类来包装的。
int main(int argc, char **argv) {
......省略代码
parse_command_line(argc, argv);
if (! Nodes_filename.empty())
Parent_map = build_parent_map(Nodes_filename);
if (Populate_memory)
cerr << "Loading database... ";
QuickFile db_file;
db_file.open_file(DB_filename);
if (Populate_memory)
db_file.load_file();
Database = KrakenDB(db_file.ptr());
KmerScanner::set_k(Database.get_k());
QuickFile idx_file;
idx_file.open_file(Index_filename);
if (Populate_memory)
idx_file.load_file();
KrakenDBIndex db_index(idx_file.ptr());
Database.set_index(&db_index);
if (Populate_memory)
cerr << "complete." << endl;
......省略代码
for (int i = optind; i < argc; i++)
process_file(argv[i]);
......省略代码
}
这里我们可以看到QuickFile只是对文件进行了mmap映射:
void QuickFile::open_file(string filename_str, string mode, size_t size) {
......省略代码
fptr = (char *)mmap(0, filesize, PROT_READ | PROT_WRITE, m_flags, fd, 0);
if (fptr == MAP_FAILED)
err(EX_OSERR, "unable to mmap %s", filename);
valid = true;
}
并且使用load_file()函数来载入数据到内存里,其实现存在quickfile.cpp文件中:
void QuickFile::load_file() {
......省略代码
#pragma omp parallel
{
#ifdef _OPENMP
thread = omp_get_thread_num();
#endif
#pragma omp for schedule(dynamic)
for (size_t pos = 0; pos < filesize; pos += page_size) {
size_t this_page_size = filesize - pos;
if (this_page_size > page_size)
this_page_size = page_size;
memcpy(buf[thread], fptr + pos, this_page_size);
}
}
......省略代码
}
所谓的load到内存其实就是把整个文件用memcpy函数读了一遍,将数据被读入内核的page cache里。
int main(int argc, char **argv) {
......省略代码
for (int i = optind; i < argc; i++)
process_file(argv[i]);
......省略代码
}
从上面可以看到鉴定的逻辑在 process_file()中,有多少个待鉴定文件就会被process_file()处理多少遍。
void process_file(char *filename) {
......省略代码
#pragma omp parallel
{
vector<DNASequence> work_unit;
ostringstream kraken_output_ss, classified_output_ss, unclassified_output_ss;
while (reader->is_valid()) {
work_unit.clear();
size_t total_nt = 0;
#pragma omp critical(get_input)
{
while (total_nt < Work_unit_size) {
dna = reader->next_sequence();
if (! reader->is_valid())
break;
work_unit.push_back(dna);
total_nt += dna.seq.size();
}
}
if (total_nt == 0)
break;
kraken_output_ss.str("");
classified_output_ss.str("");
unclassified_output_ss.str("");
for (size_t j = 0; j < work_unit.size(); j++)
classify_sequence( work_unit[j], kraken_output_ss,
classified_output_ss, unclassified_output_ss );
......省略代码
}
从上面可以看到读取待分析序列后,将每个序列都一次性读取到work_unit这个vector里,然后又用classify_sequence对每个序列进行分析。这里,如果序列很长,work_unit这个vector占用内存也会很大,这里完全可以使用批处理的方法来优化,每个批次N条序列。
最终来到了classify_sequence函数,对序列切k-mer,然后每个k-mer通过Database.kmer_query查询到其lca_taxon_id,并且对其计数,存储在hit_counts这个map里。
void classify_sequence(DNASequence &dna, ostringstream &koss,
ostringstream &coss, ostringstream &uoss) {
......省略代码
if (dna.seq.size() >= Database.get_k()) {
KmerScanner scanner(dna.seq);
while ((kmer_ptr = scanner.next_kmer()) != NULL) {
taxon = 0;
if (scanner.ambig_kmer()) {
ambig_list.push_back(1);
}
else {
ambig_list.push_back(0);
uint32_t *val_ptr = Database.kmer_query(
Database.canonical_representation(*kmer_ptr),
¤t_bin_key,
¤t_min_pos, ¤t_max_pos
);
taxon = val_ptr ? *val_ptr : 0;
if (taxon) {
hit_counts[taxon]++;
if (Quick_mode && ++hits >= Minimum_hit_count)
break;
}
}
taxa.push_back(taxon);
}
}
uint32_t call = 0;
if (Quick_mode)
call = hits >= Minimum_hit_count ? taxon : 0;
else
call = resolve_tree(hit_counts, Parent_map);
......省略代码
if (Quick_mode) {
koss << "Q:" << hits;
}
else {
if (taxa.empty())
koss << "0:0";
else
koss << hitlist_string(taxa, ambig_list);
}
koss << endl;
}
然后通过resolve_tree计算得到权重和最大的leaf-to-root path,这就是最终的鉴定结果。
call = resolve_tree(hit_counts, Parent_map);
鉴定结果的输出在hitlist_string函数中来实现
koss << hitlist_string(taxa, ambig_list);
至此,整个鉴定流程完毕。
槽点
- 研究源码的过程中,邪恶的Perl和Shell代码很难阅读,尤其是Perl代码完全是天书,非常不利于代码的传播和改进。编译型语言来负责计算密集型操作,解释性语言来负责其他部分,这应该是一个较好的组合。未来可以用Python来代替Perl和Shell。
- 对于待分析序列可以使用批处理的方式来节省内存,不过相对于已经使用了200G左右内存的数据库和索引,这点内存似乎可以忽略。
- 代码风格有点乱。
对最初始问题的解答
Q:为什么Kraken的分析速度那么快?
数据库使用mmap加载到内存,并且使用了索引,速度很快。当然,建库的过程很慢。
Q:为什么kraken的数据库有几百G那么大?
由参考序列的大小决定,并且由于是用k-mer->tax_id的建库方式,所以数据库的大小跟参考序列和k-mer长度成线性关系。
为什么Kraken建库的速度非常慢?
通过上述建库过程的解读应该知道耗时的过程在 数据库排序和创建索引,还有遍历设置lca这几个步骤,确实很需要时间。
Q:为什么Kraken数据库的载入速度非常慢?
载入的过程其实就是用memcpy对已经mmap了的数据库文件进行了遍历读操作,所以,数据库文件有多大,载入速度就有多长,成线性关系。
Q:Kraken的数据库能否拆分使得其能够分布式运行?
可以拆分,只是原生代码不支持,需要自己修改代码来实现。因为,k-mer->tax_id这样的数据库结构,我们知道如果拆分出来多个数据库后,可以分别对其做索引,然后分布在多台服务器上。在每台服务器上,将待鉴定序列分别去query本机的数据库[Map],得到hit_counts,然后在一台服务器上合并hit_counts[Reduce],此数据量已经不大了,然后再对其做resolve_tree即可得到最终结果。通过此方法我们可以实现MapReduce的架构,当有大批量序列需要鉴定时可以获得很高的生产效率,而且可以避免对超大内存服务器的硬需求。